
Par Philippe GARCELON
Cet article s’inspire du travail effectué par les chercheurs, David Porco et Louis Deharveng (Museum National d’Histoire Naturelle) qui exploitent l’outil génétique et ont eu l’amabilité de me faire parvenir des documents relatifs à leurs recherches sur les collemboles.
Introduction
Malgré l’apparition des techniques d’identification génétique, sur la base du code-barre ADN, les biologistes définissent encore majoritairement les espèces à l’aide de clefs de détermination issues de l’observation de leurs caractéristiques communes, en partant des plus générales aux plus particulières. Ce travail nécessite la collecte de spécimens vivants ou suffisamment bien conservés pour permettre de lister de manière exhaustive leurs détails morphologiques.
Actuellement, le recours au code barre ADN est essentiellement le fait de laboratoires spécialisés en génétique. Les nouvelles espèces font donc encore et prioritairement, l’objet de descriptions morphologiques détaillées. Cependant apparaissent progressivement des études sur le code barre ADN qui impactent sensiblement le travail des taxonomistes.
NB : A la suite de cet article, vous trouverez quelques compléments d’information qui, sans être indispensables à sa lecture, permettent une meilleure compréhension de ce dernier.
Taxonomie « traditionnelle »
Je reprends ici une description des particularités morphologiques d’une nouvelle espèce de collembole Deutonura gibbosa (Porco, Bedos & Deharveng, 2010) appartenant à la sous-famille des Neanurinae, au sein de la famille des Neanuridea (Poduromorphe).

La taxonomie « traditionnelle » répertorie chaque détail morphologique sous forme d’un inventaire comprenant des dessins et un tableau de synthèse utilisant une terminologie abrégée spécifique. Dans le cas présent (Deutonura gibbosa sp.nov), les « poils » sont identifiés selon :
- Leur nature : ML (macroseta long), MC (macroseta court), Me (mesosetae), Mi (microsetae)
- Leur implantation : I (thorax, segments I), Abd.4 (abdomen, segment IV), Ant.2 (antenne, second segment) etc…
- Leur nombre
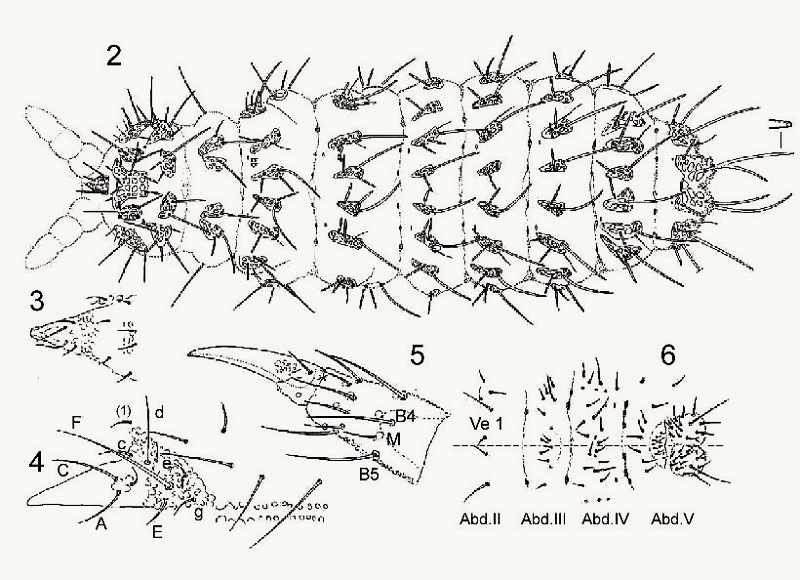
Sur la planche qui suit, différentes parties du corps sont représentées pour y répertorier l’implantation des poils :
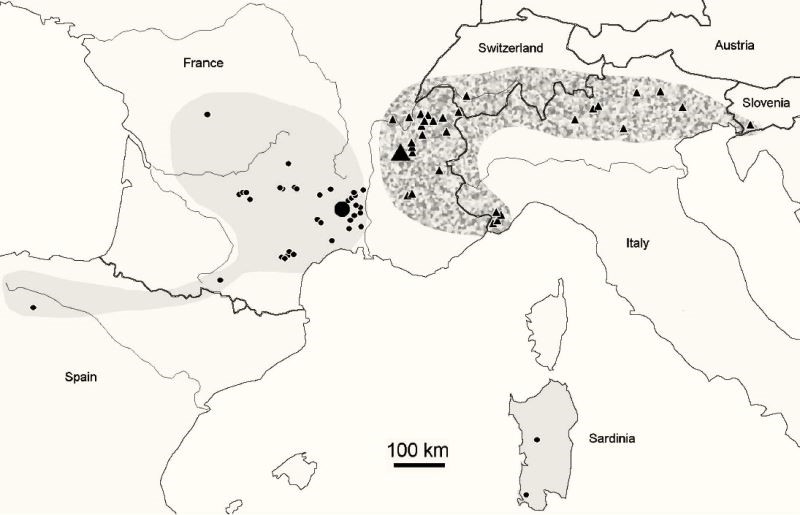
On trouve également, pour accompagner ces descriptions des cartes montrant les répartitions géographiques des espèces, généralement accompagnée de la liste des lieux et le nombre de spécimens qui y sont prélevés.
Diversité cryptique
Si documentées soient-elles, ces clefs d’identification demeurent pourtant incomplètes, car elles ne permettent pas de mettre en évidence certaines diversités, autres que morphologiques. On parle alors d’une diversité cryptique, caractérisée par une spécificité génétique pour des spécimens indissociables au niveau morphologique. Cette diversité fut mise en évidence lors d’une étude (James et al. – 2010) qui montre que la description du Lumbricus terrestris (ver de terre) s’appliquait en fait à deux espèces distinctes Lumbricus terrestris et Lumbricus herculeus. Decaëns al. (2013) estime que 30% en moyenne des espèces définies selon leur morphologie seraient des complexes d’espèces cryptiques. Une autre étude (Janzen et al. 2013) montre que 32 espèces de papillons Saturnidae, décrites morphologiquement, représentent en réalité 49 espèces distinctes au plan génétique. Ces seuls exemples illustrent ce nouveau risque que l’on nomme « chimère biologique » qui réduirait plusieurs espèces véritables à une dénomination unique.
Code-barre ADN – séquençage génétique
Afin de pouvoir aller plus loin dans l’approche taxonomique, les chercheurs ont donc abordé cette question sur le plan de la génétique. A cette occasion, ils ont remarqué qu’il n’était pas nécessaire de séquencer la totalité de la chaîne d’ADN d’un spécimen pour pouvoir discriminer son espèce. En effet, Ils ont découvert que l’analyse de portions (séquences) de cette longue chaîne était suffisante (ci-dessous) :
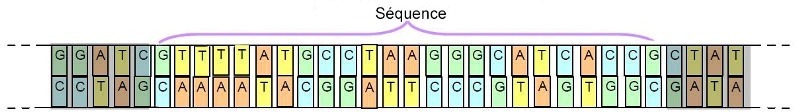
Important : Pour définir cette séquence, il faut cependant en identifier une qui varie d’une espèce à l’autre, tout en restant stable pour une même espèce.
Depuis le milieu des années 1980, les bactériologues ont utilisé le séquençage génétique dans le cadre de leurs diagnostics des infections bactériennes (légionellose, certaines formes de méningite, etc.). Le gène séquencé étant l’ARN ribosomique 16S. Pour en savoir plus, cliquer ici (téléchargement PDF).
En 2003, Paul Herbert (Directeur du Biodiversity Institute of Ontario) et son équipe, axent leurs recherches sur le séquençage d’un gène (portion définie de l’ADN qui correspond à une unité d’hérédité de base) différent, provenant d’une région spécifique de l’ADN présent dans le génome mitochondrial des cellules eucaryotes (voir compléments d’information à la suite de cet article). Cette portion d’ADN devenue aujourd’hui un standard d’identification correspond à une partie du gène qui code la « sous-unité 1 de la cytochrome C Oxydase », une protéine nommée CO1 (il constitue une portion du code d’une enzyme « Cytochrome c Oxydase » indispensable à la biochimie de la chaîne respiratoire).
Le gène CO1 choisi par Herber, tout en répondant à la nécessité première évoquée précédemment, présente aussi d’autres avantages, comme par exemple sa vitesse d’évolution élevée et le fait qu’il permet une accumulation appréciable de mutation au sein d’une espèce. Il apparaît, par comparaisons de séquences ADN, que le gène CO1 diverge suffisamment pour permettre une discrimination des espèces, voire la discrimination de populations au sein d’une même espèce (Cox and Hebert 2001- Wares and Cunningham 2001).
En outre, comme il est déjà très présent au sein de la cellule qui compte entre 300 et 2000 mitochondries et également sous forme de copies (de 5 à 10) dans la mitochondrie elle-même, il est relativement facile à multiplier, par une technique biochimique complexe nommée « Amplification PCR » (Polymerase Chain Reaction).
Ce procédé permet l’obtention un très grand nombre de copies (100 à 10 000) d’une séquence donnée d’ADN afin de disposer d’une quantité suffisante permettant aux machines d’assurer le séquençage.
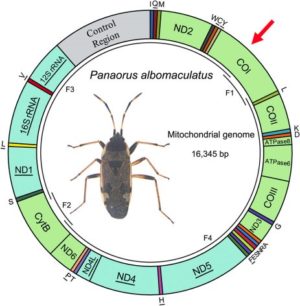
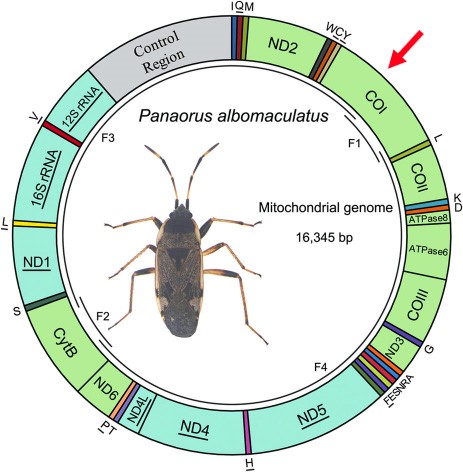
Ci-contre, exemple d’une carte du génome mitochondrial d’un hémiptère sur laquelle on distingue des divisions, correspondant chacune à un gène. Dans le cas de cet insecte, 13 de ces gènes sont codants pour la protéine (c’est le cas de CO1 marqué d’une flèche rouge).
Le genre Deutonura qui compte plus de 56 espèces est un des principaux représentant de la grande sous-famille Neanurinea qui en compte au total 784 (dernière actualisation au 28-03-2018). Si certaines espèces comme Deutonura phleagea (Caroli, 1912- Cassagnau, 1979) possèdent une large aire de répartition, la majeure partie d’entre elles été observées dans des zones bien plus restreintes, certaines mêmes étant des espèces endémiques étroites. La répartition géographique de Deutonera gibbosa sp.nov, espèce très commune du genre, couvre les Alpes et le sud du Jura.
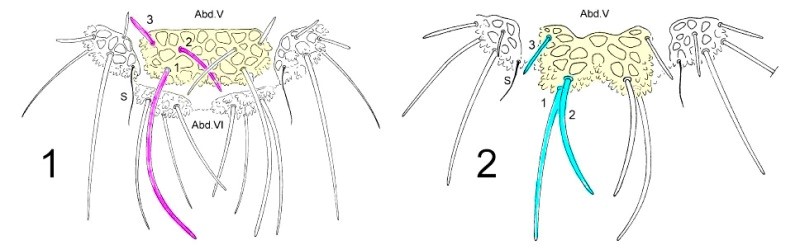
Pour dissocier les espèces du genre Deutonera, la taxonomie classique prend principalement en compte l’arrangement des tubercules dorsaux ainsi que le nombre de poils sur chaque tubercule. L’observation des deux espèces, Deutonura deficiens sylvatica (fig.1) et Deutonura gibbosa sp.nov. (fig.2) montre cependant une implantation similaire des poils dorsaux et un même nombre par tubercules. (J’ai colorisé les croquis et corrélé les couleurs avec les tableau I et II des distances )
La seule différence constatée, comme le montre le croquis ci-dessous, réside en une bilobation et un allongement du tubercule dorso-interne sur Abd.V (en jaune), associés à une modification de l’arrangement des poils (rose/bleu) et de leurs longueurs sur ce même tubercule.
Les chercheurs ont effectué un séquençage sur 17 spécimens de 5 espèces du genre Deutonura (6 D. caerulescnens, 5 D. deficiens sylvatica, 1 D. decolorata, 4 D. gibbosa sp.nov. et 1 D. vallespirensis).
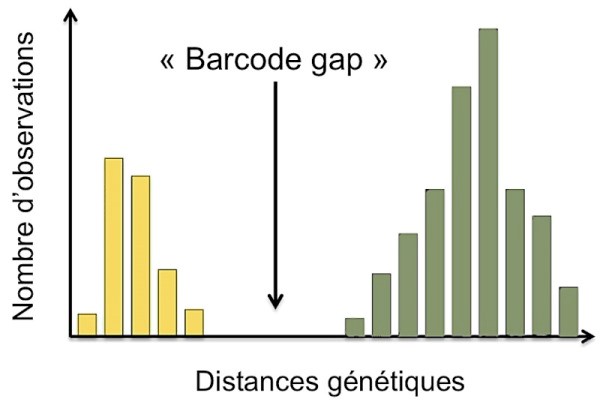
Définition : La distance génétique est un indice (valeur numérique) proportionnel au nombre de différences qui existent entre deux séquences d’un même fragment de gène et qui peut prendre en compte différents modèles d’évolution. Par exemple, l’illustration ci-contre montre une différence significative des distances génétiques, en jaune pour des individus d’une même espèce (intra-spécifique) et en vert pour des individus appartenant à des espèces différentes (inter-spécifique).
Les distances génétiques mesurées sont reportées sur les tableaux ci-dessous : Tableau.1 (interspécifiques), Tableau.2 (intraspécifiques).

Ces valeurs sont également illustrées par l’arbre ci-dessous, généré à partir d’un modèle de transcription des nucléotides nommé « K2P » (Kimura deux paramètres) sur lequel les côtés supérieurs et inférieurs des triangles sombres font apparaître respectivement les distances génétiques maximales et minimales au sein des espèces.
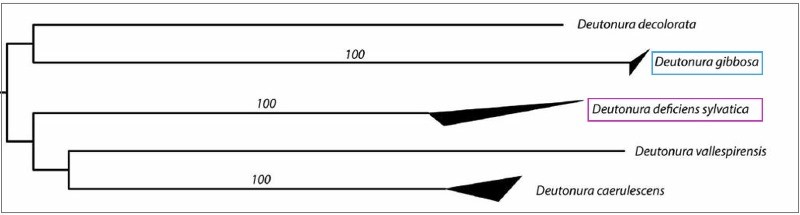
On visualise effectivement (en bleu) que Deutonura gibbosa sp.nov. (Barcoding effectuée avec 4 spécimens), possède la plus faible valeur intraspécifique (0.0006) du genre Deutonura, alors que pour son plus proche voisin (en rose) Deutonura deficiens sylvatica, la mesure effectuée à partir des 5 spécimens, montre une distance interspécifique de 0,2074.
Ces deux valeurs intra et interspécifiques, ainsi que les distances caractérisant les autres espèces de ce même genre (Deutonura) apportent un support génétique permettant de délimiter Deutonura gibbosa sp.nov en tant que lignée mitochondriale distincte.
Conlusion
Comme le souligne cet exemple, les différences morphologiques minimes entre deux espèces D. gibbosa sp. nov. et D. deficiens sylvatica sont nettement plus marquées sur le plan génétique. Ce constat effectué dès les années 2010 avec lequel convergent des études plus récentes confirme le potentiel de l’outil génétique en ce qui concerne la caractérisation des espèces. Plus largement, de très nombreuses espèces non encore décrites ou qui nécessitent des examens approfondis bénéficient de cet apport qui accroît fiabilité et précision. Mais les ressources du code-barre ADN s’étendent aussi bien au-delà de l’identification des espèces en impactant de manière plus générale l’ensemble du champ de la biodiversité.
Le code-barre ADN qui peut être reproduit selon les besoins permet des analyses massives avec un gain de temps considérable comparé à l’identification morphologique dont on vient d’entrevoir certaines limites. L’utilisation du code barre ADN apparaît donc aujourd’hui comme un outil qui élargit considérablement le champ d’investigation des biologistes. Alors que la taxonomie classique nécessite d’étudier de spécimens intacts, l’étude génétique s’accommode de divers états comme par exemple les œufs ou les larves mais aussi des fragments de cadavres, d’excréments, de mucus ou d’autres traces présentes dans divers substrats (on parle à ce propos d’analyse d’ADN intracellulaire et d’analyse d’ADN extracellulaire). Ainsi, l’analyse de l’eau d’une marre peut révéler qu’une espèce donnée de grenouille y a évoluée, même si cette grenouille n’a pu y être physiquement prélevée.
Le Code-barre ADN ouvre également la voie à la reconstitution historique, tant au niveau des mécanismes de séparation de deux espèces que dans l’évolution de leur répartition géographique (phylogéographie). A ce propos, D. Porco et al. (2013) ont montré comment des collemboles européens introduits en Amérique du Nord y sont devenus invasifs.
L’utilisation du code-barre ADN permet en outre une mise en évidence des comportements prédateurs de certaines espèces à travers l’analyse de leurs régimes alimentaires, par prélèvement des contenus de tubes digestifs ou des déjections. Ces études donnent un nouvel éclairage sur les interactions proies/prédateurs ou hôtes/parasites (lien).
Cette technique a cependant ses propres limites. Effectivement, les chercheurs ont parfois constaté des similitudes génétiques sur des spécimens morphologiquement distincts ou, pour des populations géographiquement isolées où une même espèce peut présenter une forte variabilité génétique. Pour conforter leurs conclusions, les scientifiques ont donc tout intérêt à croiser les résultats du code-barre ADN avec leurs données classiques d’identification.
Complément d’informations
Séquençage ADNLa préparation du séquençage nécessite une succession d’étapes qui consiste en une destruction des tissus suivie d’une purification et d’une « amplification » réalisée sous forme d’un protocole qui régit une série d’opérations physico-chimiques. L’ADN mitochondrial est plus particulièrement utilisé pour le séquençage du génome en raison de ses propriétés intrinsèques dont, le fait qu’il est 5 à 10 fois plus spécifique que le génome de l’ADN nucléaire.

Réalisées en laboratoire par des machines automatisées, le séquençage s’effectue à partir d’un tube à essai contenant ADN, nucléotides et amorce de l’ADN polymérase (issus de l’amplification PCR évoquée plus haut). 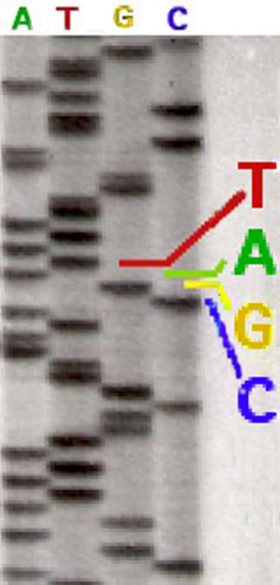 Je ne saurais détailler ici les complexes réactions de synthèse qui ont lieu dans un de ces tubes à essai. On peut juste noter que l’analyse spectrale n’est possible que grâce au marquage par des fluorochromes différents (A vert, T rouge, G jaune et C bleu), chaque couleur occupant des plages de longueurs d’onde distinctes. Lors de la restitution de l’analyse chaque nucléotide apparaît donc dans une succession correspondant à son ordre au sein de la séquence analysée.Le marquage radioactif initialement utilisé ne permettait pas de distinguer un nucléotide des trois autres. Il fallait alors séquencer à partir de quatre tubes différents, chacun possédant un type de nucléotide. Un séquençage avec marquage radioactif pouvait être lu directement à l’œil sur un gel d’acrylamide (ci-contre). De nos jours, le remplacement du marquage radioactif par des fluorochromes autorise la réalisation des quatre réactions au sein d’un même tube. Les séquenceurs modernes détectent les intensités de fluorescence et possèdent des logiciels qui permettent de traduire les résultats sous forme graphique. La position des nucléotides (A, G, T, C) nommée séquence caractérise l’information génétique. On remarque sur la partie supérieure, la succession des nucléotides repérés par leur lettre et accompagnées de chiffres qui permettent de les situer au sein de la séquence étudiée. Sur la partie inférieur, les courbes de quatre couleurs correspondent à l’amplitude (intensité) des fluorescences mesurées pour chacun des marqueurs. Au-delà de ce type de présentation, toutes les informations sont également consignées sous formes de tableaux utilisant une nomenclature internationale qui permet des comparaisons automatisées avec les bases de données existantes. |
Bases de donnéesLes bases de données génétiques deviennent cruciales en biologie, de ce fait la publication d’un article scientifique décrivant une séquence biologique est assujettie au dépôt préalable de ses données génétiques sur une des trois bases suivantes :
Le nombre de séquence ou de génomes entiers qui y sont déposés s’est accru de manière exponentielle entre 2000 et 2014 alors que parallèlement les coûts des séquençages diminuaient dans des proportions similaires facilitant d’autant les recours à ce type de techniques. Aujourd’hui, il existe des centaines de bases de données, ce qui ont conduit à la création d’une base de données des bases de données : NAR database A titre indicatif, en septembre 2012 les bases de données nucléiques comptaient, pour les seuls invertébrés, 31.907.138 entrées, soit un total de 52.527.673.643 de nucléotides ! Dans le cas spécifique des code-Barre ADN, il existe par exemple une base de données gérée par International Barcode of Life qui au 02/05/2018 disposait dans sa plateforme bio-informatique BOLD d’un ensemble de plus de 6 millions de codes-barre ADN dont, pour le règne animal, 4.367.942 codes-barres représentant 154 271 espèces et issus de 238 pays. Cette plateforme dispose en outre d’outils permettant de comparer la séquence ADN d’un organisme inconnu à l’ensembles des code-barre ADN répertoriés dans la base. |
Sources :
- Porco D. ; Bedos A. & Deharveng L. (2010): Description and DNA barcoding assessment of the new species Deutonura gibbosa (Collembola: Neanuridae: Neanurinae), a common springtail of Alps and Jura. Zootaxa 2639, 59-68 (lien)
- « Amplification PCR- Ifremer » : http://www.bibliomer.com/
- Société Française d’Ecologie et d’Evolution : Article : « Le barcoding ADN » (T. Decaëns, D. Porco & R. Rougerie)
- Site internet sur les collemboles (Phjilippe Garcelon) : https://collemboles.fr
Thank You